
This project was to re-develop a faster data processing engine for Mass Spectrometry data for researchers in proteomics researching crosslinked peptide pairs.
Technologies: Python, Numpy
The team based at the University of Edinburgh and Technical University of Berlin are global leaders in the field and had already developed their own data processing system. We were asked to redevelop the system with two outcomes, make the data processing faster and create a process template to open up software development silos within the research teams.
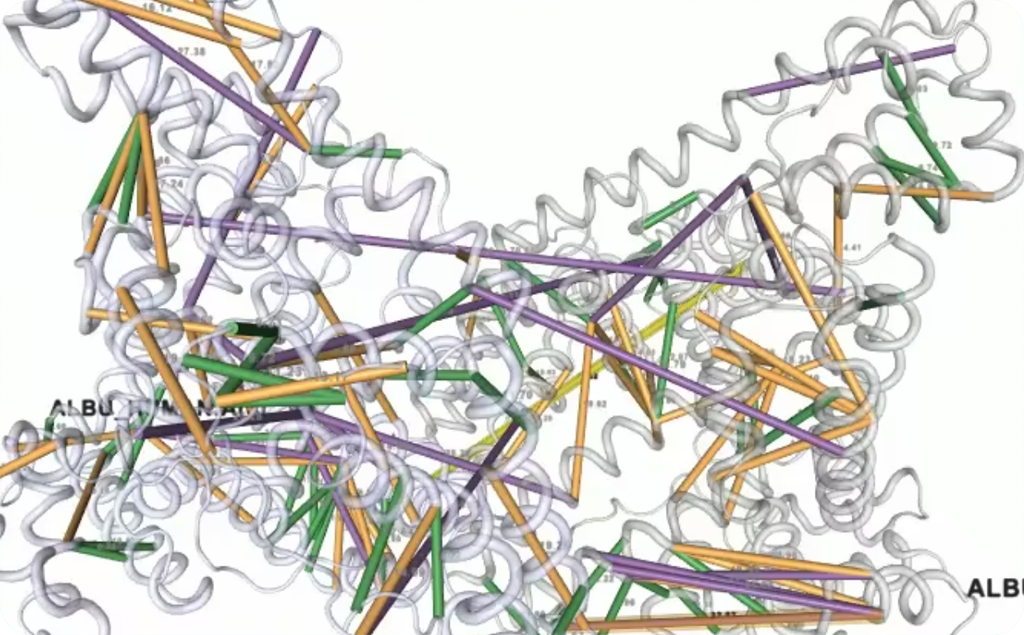
The science: Crosslinking Protein Research, both the structure of proteins and interactions between proteins and other molecules are studied. This is done by adding special chemicals called crosslinkers to a sample, cutting the proteins in that sample into smaller pieces, called peptides, and analysing those peptides in a mass spectroscope. Deriving the candidate peptides, and therefore the corresponding proteins, from the data produced by the mass spectroscope is a computationally complex problem: by calculating all potential spectra for all expected peptides we can match, through a statistical process, the mass spectroscope data to peptides and from that derive what parts of a protein, or which proteins, were a set distance apart at the time sample was prepared, and thus this provides information about the 3D structure of the protein or the interaction between proteins. The current software produced by the leading research teams in the field from two research institutes was the best available tool and they asked us to rewrite and improve it.
The software helps a wider range of researchers use, work with and contribute to the software. The target outcomes for more efficient processing have also been met.